Réseaux de co-expression de gènes
L'expression d'un gène est la transcription et la traduction d'un gène en ARN messager et donc en protéines (sauf cas des micro ARN). Il y a co-expression quand plusieurs gènes s'expriment dans des conditions similaires. Un réseau de co-expression de gène (GCN) est un graphe, où chaque nœud correspond à un gène et où une paire de nœuds est reliée par un arc s'il existe une relation significative de co-expression entre eux[1]. Un réseau de co-expression de gène peut être construit, si l'on dispose de suffisamment de profils d'expression de gènes, en provenance de plusieurs échantillons ou d'expérimentations, en recherchant des paires de gènes qui ont un modèle d'expression similaire. C'est-à-dire un modèle d'expression où les niveaux de transcription de deux gènes exprimés conjointement montent et retombent simultanément dans les différents échantillons. Les réseaux de co-expression de gènes (GCN) sont intéressants sur le plan biologique car ils mettent en évidence les gènes qui sont contrôlés par le même programme de régulation transcriptionnel, ou alors qui sont fonctionnellement liés, ou bien encore qui sont des membres du même réseau de régulation génétique[2].
La direction et le type de la relation de co-expression ne sont pas définis dans les réseaux de co-expression de gènes, au contraire d'un réseau de régulation génique (GRN), où un arc orienté reliant deux gènes représente un processus biochimique comme une réaction, une transformation, une interaction, une activation ou une inhibition[3]. Par rapport à un GRN, un GCN ne permet pas de déduire les relations de causalité entre les gènes et dans un GCN les arcs indiquent seulement une corrélation d'expression de ces différents gènes[4]. Les modules ou les sous-graphes fortement interconnectés dans les réseaux de co-expression de gène (GCN) correspondent aux groupes de gènes ayant une fonction similaire ou participant à un processus biologique commun[3].
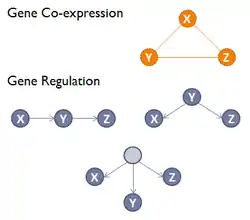
Les réseaux de la co-expression de gènes sont généralement construits à l'aide d'ensembles de données générées par l'expression de gènes au moyen de technologies à haut débit telles que les biopuces/micromatrices (microarray)ou RNA-Seq.
Histoire
Le concept de réseaux de co-expression de gènes a été introduit par Butte et Kohane en 1999 en tant que "relevance networks"[5]. Bute et Kohane intégrèrent cette approche plus tard avec des données d'expression de gènes pour construire le premier réseau de co-expression de gènes[6].
Construction d'un réseau de co-expression de gènes
Pour construire un réseau de co-expression de gènes, il faut suivre une approche en deux étapes : d'abord calculer le degré de co-expression, puis sélectionner le seuil à partir duquel on considère que cette expression devient significative.
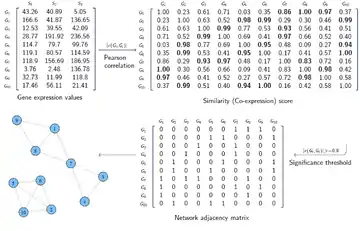
Les données d'entrée pour la construction d'un réseau de co-expression de gènes sont souvent représentées par une matrice. Si nous avons les valeurs d'expression génique de m gènes pour n échantillons (conditions), les données d'entrée seraient une matrice m × n, appelée matrice d'expression. Par exemple, dans une expérience de microréseau, les valeurs d'expression de milliers de gènes sont mesurées pour plusieurs échantillons. Dans la première étape, un score de similarité (mesure de co-expression) est calculé pour chaque paire de lignes dans la matrice d'expression. La matrice résultante serait une matrice m × m, appelée matrice de similarité. Chaque élément de cette matrice montre à quel point le niveau d'expression de deux gènes est similaire. Dans la deuxième étape, les éléments de la matrice de similarité qui dépassent un certain seuil (c.-à-d. des co-expressions significatives) sont remplacés par 1 et les éléments restants sont remplacés par 0. La matrice résultante, appelée matrice adjacente, représente le graphe du réseau. Dans cette matrice, chaque élément montre si deux gènes sont connectés dans le réseau (les éléments 1) ou non (les éléments 0).
Le coefficient de corrélation de Pearson (en), l'information mutuelle, la corrélation de Spearman et la distance euclidienne sont les quatre méthodes les plus fréquemment utilisés pour construire des réseaux de co-expression de gènes. Plusieurs autres mesures, telles que la corrélation partielle[7], la régression[8], et une combinaison de corrélation partielle et échange d'informations[9] ont aussi été utilisés.
Voir aussi
- Analyse de réseaux par corrélation pondérée (en)
- réseaux de régulation de l'expression génique (en) ; Régulation de l'expression des gènes
- Inférence d'un réseau biologique (en)
- réseau biologique
Références
- Joshua M Stuart, Eran Segal, Daphne Koller et Stuart K Kim, « A gene-coexpression network for global discovery of conserved genetic modules », Science, vol. 302, no 5643, , p. 249–55 (PMID 12934013, DOI 10.1126/science.1087447, Bibcode 2003Sci...302..249S)
- Matthew T Weirauch, « Gene coexpression networks for the analysis of DNA microarray data », Applied Statistics for Network Biology: Methods in Systems Biology,
- Swarup Roy, Dhruba K Bhattacharyya et Jugal K Kalita, « Reconstruction of gene co-expression network from microarray data using local expression patterns », BMC Bioinformatics, vol. 15, , S10 (DOI 10.1186/1471-2105-15-s7-s10)
- Riet De Smet et Kathleen Marchal, « Advantages and limitations of current network inference methods », Nature Reviews Microbiology, vol. 8, no 10, (DOI 10.1038/nrmicro2419)
- Atul J Butte et Isaac S Kohane, « Unsupervised knowledge discovery in medical databases using relevance networks », Proceedings of the AMIA Symposium,
- Atul J Butte et Isaac S Kohane, « Mutual information relevance networks: functional genomic clustering using pairwise entropy measurements », Pac Symp Biocomput, vol. 5,
- Nathalie Villa-Vialaneix, Laurence Liaubet, Thibault Laurent, Pierre Cherel, Adrien Gamot et Magali SanCristobal, « The structure of a gene co-expression network reveals biological functions underlying eQTLs », PLOS ONE, vol. 8, no 4, , p. 60045 (DOI 10.1371/journal.pone.0060045, Bibcode 2013PLoSO...860045V)
- Staffan Persson, Hairong Wei, Jennifer Milne, Grier P Page et Christopher R Somerville, « Identification of genes required for cellulose synthesis by regression analysis of public microarray data sets », Proceedings of the National Academy of Sciences of the United States of America, vol. 102, no 24, , p. 8633–8 (PMID 15932943, DOI 10.1073/pnas.0503392102, Bibcode 2005PNAS..102.8633P)
- Antonio Reverter et Eva KF Chan, « Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks », Bioinformatics, vol. 24, no 21, , p. 2491–2497 (DOI 10.1093/bioinformatics/btn482)