Pneumonie interstitielle aigüe
La pneumonie interstitielle aigüe est une forme fulminante de pneumopathie interstitielle idiopathique[1]. Cette maladie pulmonaire grave et rare affecte généralement des individus par ailleurs en bonne santé. Elle n'a pas de cause connue ni de remède.
| Spécialité | Pneumologie |
|---|
| CIM-10 | J84.1 |
|---|---|
| CIM-9 | 516.3 |
| OMIM | 178500 |
| DiseasesDB | 4815 |
| MeSH | D011658 |
![]() Mise en garde médicale
Mise en garde médicale
La pneumonie interstitielle aigüe est souvent classée à la fois comme une maladie pulmonaire interstitielle et comme une forme de syndrome de détresse respiratoire aigüe (SDRA), mais elle se distingue des formes chroniques de pneumonie interstitielle telles que la fibrose pulmonaire idiopathique[2].
Symptômes
Les symptômes les plus courants de la pneumonie interstitielle aigüe sont une toux très productive avec expectoration de mucus épais, de la fièvre et des difficultés respiratoires. Ceux-ci se produisent souvent sur une période d'une à deux semaines avant de consulter un médecin. La présence de mucosité explique que la personne ressent une sensation de suffocation de quasi-noyade. Les difficultés respiratoires peuvent rapidement évoluer vers une incapacité à respirer sans assistance (insuffisance respiratoire).
La pneumonie interstitielle aigüe progresse généralement rapidement. L'hospitalisation et la ventilation mécanique sont souvent nécessaires quelques jours à quelques semaines seulement après l'apparition des premiers symptômes.
Diagnostic
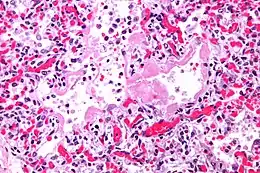
La progression rapide des symptômes initiaux à l'insuffisance respiratoire est une caractéristique clé. Le diagnostic s'appuie sur une tomodensitométrie (TDM) permettant d'écarter les autres causes de lésion aigüe du poumon[3]. Une biopsie du poumon qui montre l'organisation de lésions alvéolaires diffuses peut aussi être pratiquée. D'autres tests de diagnostic sont utiles pour exclure d'autres conditions similaires, mais l'histoire, la radiographie et la biopsie sont essentielles. Ces autres tests peuvent inclure des analyses sanguines de base, des hémocultures et un lavage bronchoalvéolaire.
Le tableau clinique est similaire à celui du SDRA, mais sa cause reste inconnue.
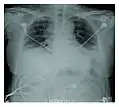 Radiographie montrant une réduction marquée de la capacité pulmonaire
Radiographie montrant une réduction marquée de la capacité pulmonaire_Idiopathic_DAD_3.jpg.webp)
_Idiopathic_DAD_2.jpg.webp)
_Idiopathic_DAD.jpg.webp)
Traitement
Le traitement est principalement de soutien. La gestion dans une unité de soins intensifs est nécessaire et le besoin de ventilation mécanique est courant. La thérapie avec des corticostéroïdes est généralement tentée, bien que leur utilité n'ait pas été établie. Le seul traitement qui a réussi jusqu'à présent est une greffe de poumon.
Pronostic
Soixante pour cent des personnes atteintes de pneumonie interstitielle aigüe meurent au cours des six premiers mois de la maladie[4]. La survie médiane est d'un mois et demi.
Cependant, la plupart des gens qui ont subi un épisode n'en ont pas de second. Les personnes qui survivent récupèrent souvent complètement leur fonction pulmonaire.
Épidémiologie
La pneumonie interstitielle aigüe survient le plus souvent chez les personnes de plus de quarante ans. Elle affecte également hommes ou femmes. Il n'y a aucun facteur de risque identifié ; en particulier, le tabagisme n'est pas associé à un risque accru.
Histoire
La pneumonie interstitielle aigüe a été décrite pour la première fois en 1935 par Louis Hamman et Arnold Rich, et a reçu le nom de syndrome de Hamman-Rich[5].
Notes et références
- Orphanet - Pneumopathie interstitielle aigüe
- Hamman L. et Rich A.R., « Acute diffuse interstitial fibrosis of the lungs », Bull. Johns Hopkins Hosp., vol. 74, , p. 177–212
- Manuel MSD — Pneumonie interstitielle aigüe
- D Bouros, Nicholson AC, Polychronopoulos V et du Bois RM, « Acute interstitial pneumonia », Eur. Respir. J., vol. 15, no 2, , p. 412–8 (PMID 10706513, DOI 10.1034/j.1399-3003.2000.15b31.x, lire en ligne)
- L Hamman et Rich AR, « Fulminating diffuse interstitial fibrosis of the lungs », European Respiratory Society, vol. 51, , p. 154–163 (PMID 21407504)