Maladie de moyamoya
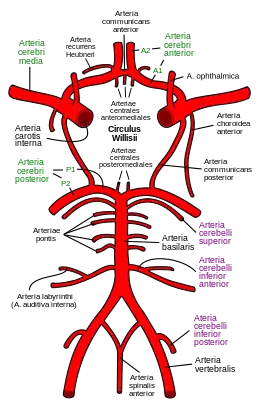
La maladie de moyamoya (appelée aussi moyamoya, maladie de Nishimoto, maladie de Nishimoto-Takeuchi-Kudo ou maladie obstructive du polygone de Willis) est une maladie vasculaire cérébrale chronique caractérisée par la sténose et l’occlusion progressive de la terminaison des artères carotides internes intracrâniennes et de la partie proximale des artères du polygone de Willis.
| Spécialité | Neurologie |
|---|
| CIM-10 | I67.5 |
|---|---|
| CIM-9 | 437.5 |
| OMIM | 252350 607151 608796 |
| DiseasesDB | 8384 |
| eMedicine | 1180952 |
| MeSH | D009072 |
| Patient UK | Moyamoya-Disease |
![]() Mise en garde médicale
Mise en garde médicale
En réaction à cette occlusion se développe un réseau vasculaire anormal au niveau de la base du crâne, dont l’aspect angiographique en « nuage de fumée » (en japonais : もやもや, moyamoya) a donné son nom à la maladie.
Historique
Une technique d'angiographie carotidienne percutanée, imaginée par Shimizu en 1937, a été popularisée auprès des neurochirurgiens au Japon peu après la seconde guerre mondiale[1].
En 1957, Takeuchi et Shimizu ont signalé un cas d'une maladie inconnue caractérisée par une hypoplasie des artères carotides internes.
En 1968 et 1969, Kudo, Nishimoto et Takeuchi, ainsi que Suzuki et Takaku, ont publié leurs études dans la littérature anglaise, ce qui a grandement contribué à la reconnaissance de la maladie de moyamoya dans le monde entier[1]. Ces auteurs ont également décrit les 6 étapes de progression angiographique, du stade 1 (rétrécissement de l'artère carotide) au stade 6 lorsque se développe un réseau vasculaire anormal au niveau de la base du crâne avec son l’aspect angiographique en « nuage de fumée ».
Épidémiologie
Cette maladie est responsable de 6 % de l’ensemble des accidents vasculaires cérébraux (AVC) de l’enfant[2].
La maladie de moyamoya s’observe surtout en Asie, et plus particulièrement au Japon. Dans ce dernier pays, le nombre annuel total de patients traités pour la maladie de moyamoya a été estimé à 3 900 en 1994, avec un taux de prévalence et d'incidence respectivement de 3,16 et 0,35 pour 100 000 habitants[3]. En Europe ou aux États-Unis, la maladie de moyamoya serait 20 fois moins fréquente. On pourrait l’estimer à environ 0,3 cas pour 100 000 habitants en France[2].
Au Japon le sex ratio (femmes/hommes) est de 1,8[3]. Les formes familiales représentent environ 10 % des cas au Japon[3], elles sont exceptionnelles en Europe[2].
Au Japon, un pic de distribution des âges des patients est observé pour des valeurs de 10 à 14 ans. L'âge de début de la maladie se situe avant 10 ans pour 47,8 % des patients mais certains d'entre eux ont développé la maladie à l'âge de 25–49 ans[3]. Dans ce dernier cas, il existe souvent une cause autre (athérome, drépanocytose, séquelles de radiothérapie...). On parle alors plutôt de « syndrome de moyamoya », par opposition à la « maladie de moyamoya » des formes infantiles[4].
Physiopathologie
Il existe une participation génétique, une mutation sur le gène RNF213 étant retrouvée[5]. Le syndrome d'Alagille est également associé des anomalies vasculaires intra-cérébrales du type moyamoya[6].
Clinique
La plupart des symptômes s’expliquent par l’irrigation sanguine insuffisante au niveau du cerveau. Certaines zones cérébrales ne reçoivent pas assez d’oxygène pour fonctionner normalement. Les symptômes dépendent de la zone touchée : troubles de la vision, de la motricité, de la sensibilité... La répétition des épisodes de « privation » d’oxygène peut endommager définitivement les zones touchées, ce qui explique que certaines séquelles soient permanente. L'atteinte peut être bilatérale dans 40 % des cas[7]. Une hyperventilation peut favoriser l'apparition des symptômes[8].
Examen complémentaire
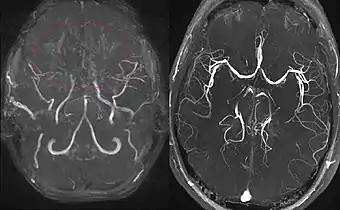
(à droite) Patient en bonne santé, pour comparaison.
L'IRM cérébrale constitue l'examen de choix chez l'enfant, permettant de faire le diagnostic de l'accident vasculaire cérébral et d'en suspecter le mécanisme[9]. l'examen doit être complété par une angiographie cérébrale au cours d'un scanner.
Traitement
La revascularisation chirurgicale reste le traitement principal[10], permettant de réduire le risque de récidive d'un accident vasculaire cérébral[11]. La mise sous antiagrégant plaquettaire est recommandée.
Notes et références
- (en) Oshima H, Katayama Y, « Discovery of cerebrovascular moyamoya disease: research during the late 1950s and early 1960s », Childs Nerv Syst, vol. 28, no 4, , p. 497-500. (PMID 22327249, DOI 10.1007/s00381-012-1708-x)
- M. Carneiro, « La maladie de moyamoya pédiatrique | Centre National de Référence de l'AVC de l'enfant », sur www.chu-st-etienne.fr, (consulté le ).
- (en) Wakai K., Tamakoshi A., Ikezaki K., Fukui M., Kawamura T., Aoki R., Kojima M., Lin Y., Ohno Y., « Epidemiological features of moyamoya disease in Japan: findings from a nationwide survey », Clinical neurology and neurosurgery, vol. 99, no Suppl 2, , S1-5 (PMID 9409395, résumé)
- Arias EJ, Derdeyn CP, Dacey RG, Zipfel GJ, Advances and surgical considerations in the treatment of Moyamoya disease, Neurosurgery, 2014;74:S116–S125
- (en) Liu W, Morito D, Takashima S et al., « Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development », PLoS One, vol. 6, no 7, , e22542. (PMID 21799892, PMCID PMC3140517, DOI 10.1371/journal.pone.0022542, lire en ligne [html])
- (en) Candice D. Carpenter, Luke L. Linscott, James L. Leach et Sudhakar Vadivelu, « Spectrum of cerebral arterial and venous abnormalities in Alagille syndrome », Pediatric Radiology, vol. 48, no 4, , p. 602–8. (ISSN 0301-0449 et 1432-1998, DOI 10.1007/s00247-017-4043-2, lire en ligne [html], consulté le )
- Scott RM, Smith ER, Moyamoya disease and moyamoya syndrome, N Engl J Med, 2009;360:1226-1237
- Tagawa T, Naritomi H, Mimaki T, Yabuuchi H, Sawada T, Regional cerebral blood flow, clinical manifestations, and age in children with moyamoya disease, Stroke, 1987;18:906-910
- Ferriero DM, Fullerton HJ, Bernard TJ et al. Management of stroke in neonates and children: a scientific statement from the American Heart Association/American Stroke Association, Stroke, 2019;50:e51-e96
- (en) D.M. Ibrahimi, R.J. Tamargo et E.S. Ahn, « Moyamoya disease in children », Child's nervous system, vol. 26, no 10, , p. 1297-1308 (PMID 20607248, lire en ligne)
- Lee S, Rivkin MJ, Kirton A, deVeber G, Elbers J, Moyamoya disease in children: results from the International Pediatric Stroke Study, J Child Neurol, 2017;32:924-929
Voir aussi
Articles connexes
Liens externes
- Moyamoya traitment center: https://www.moyamoya.eu
- Communautés de malades atteints de maladies rares
- CERVCO : Centre de Référence des Maladies Vasculaires Rares du Cerveau et de l'Oeil, Hôpital Lariboisière, Paris : http://cervco.fr
- Association Tanguy Moya-Moya : http://www.tanguy-moya-moya.org