Achondrogenèse
L’achondrogenèse est une maladie constitutionnelle de l’os létale soit in utero soit quelques jours après la naissance. C’est la seconde maladie constitutionnelle de l’os létale après le nanisme thanatophore. L’achondrogenèse est un nanisme par raccourcissement sévère généralisé des membres ou micromélie associé à un petit tronc et un crâne anormalement grand. Il existe plusieurs types d’achondrogenèse en rapport avec des anomalies génétiques différentes :
- Type IA : Achondrogenèse type Houston-Harris
- Type IB : Achondrogenèse type Fraccaro
- Type II : Achondrogenèse type Langer-Saldino ou chondrogenèse imperfecta.
| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q77.0 |
|---|---|
| OMIM | 200600 |
| DiseasesDB | 32635 et 33398.htm 33350, 32635 et 33398 |
| MedlinePlus | 001247 |
| MeSH | C579878 |
![]() Mise en garde médicale
Mise en garde médicale
La distinction est importante en raison des modes de transmissions différents entrainant des conseils génétiques différents.
Type IB d’achondrogenèse type Fraccaro
Étiologie
Mutation du gène SLC26A2 solute carrier family 26 (sulfate transporter), member 2 situé sur le locus q31-q34 du chromosome 5.
La mutation de ce gène est la seule mutation génique connue responsable de cette maladie.
Incidence
Inconnue.
À la naissance
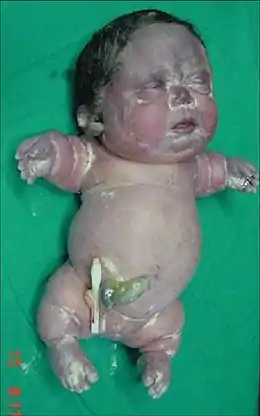
Le nouveau-né présente des membres très courts (c’est le plus sévère des nanismes). Avec des doigts et des orteils raccourcis, un abdomen protubérant et aspect gras de l’enfant par excès de tissu mou par rapport au squelette. Le crâne paraît disproportionné en comparaison au corps. Le visage est plat et le cou court. Le décès survient rapidement.
La radiographie osseuse du squelette est indispensable pour diagnostiquer avec certitude des autres maladies létales constitutionnelles de l’os.
À l’échographie
Il existe un excès de liquide amniotique (hydramnios). La biométrie des os montre que leur taille est très en dessous de la courbe de croissance normale. L’échogénicité des os est faible surtout celle de la colonne vertébrale et du crâne.
Un diagnostic précoce peut être fait dès la 10e semaine en cas d’antécédents d’achondrogenèse.
Diagnostic
La certitude du diagnostic nécessite l’étude histologique du cartilages et des os du nouveau-né.
Le diagnostic peut être fait par la recherche des mutations du gène SLC26A2. Il existe cinq mutations géniques responsable de cette maladie. Cette recherche n’est positive que dans 65 % des cas, obligeant une analyse totale du gène pour établir le diagnostic.
Diagnostic différentiel
Le diagnostic différentiel avec les autres ostéochondrodysplasies létales est parfois difficile. Un diagnostic précis nécessite :
- des radiographies précises et de bonne qualité
- du tissu permettant une étude génétique
- une biopsie cutanée permettant une culture de fibroblaste
- un échantillon du cartilage et des os pour études histologique et biochimiques
Les principales pathologies à discuter sont :
- Ostéogenèse imparfaite type II et III
- Nanisme thanatophore
- Syndrome côtes courtes Polydactylies type I, II et III
- Fibrochondrogenèse
- Achondroplasie sous la forme homozygote
Mode de transmission
Transmission autosomique récessive.
Diagnostic prénatal
L’échographie permet de détecter précocement un fœtus affecté. Le diagnostic est également possible par étude génique si les mutations des deux parents sont connues.
Conseil génétique
Les deux parents d’un fœtus ou enfant porteur de cette pathologie sont toujours porteurs d’un gène muté. Ils n’ont aucune manifestation clinique et le risque de maladie osseuse n’est pas augmenté.
Le risque d’avoir un nouveau-né affecté est de 25 %.
Type IA d’achondrogenèse ou type Houston-Farry
Étiologie
Gène inconnu.
Mode de transmission
Autosomique récessive.
Type II d’achondrogenèse ou Type Lander-Saldino ou chondrogenèse imparfaite
Étiologie
Mutation du gène COL2A1 collagen, type II, alpha 1 situé sur le locus q13.1-q13 du chromosome 12.
Cette mutation est toujours une mutation de novo.
Mode de transmission
Sources
- (fr) Site de renseignement sur les maladies rares et les médicaments orphelins
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:600972
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005