Syndrome de Beckwith-Wiedemann
Le syndrome de Beckwith-Wiedemann est une maladie génétique se caractérisant par une croissance excessive du fœtus (gigantisme), une grosse langue (macroglossie), une hémi-hyperplasie, une hypertrophie des organes (viscéromégalie), une omphalocèle ou une hernie ombilicale, une prédisposition à développer une tumeur embryonnaire, des anomalies des oreilles et des hypoglycémies.
| Spécialité | Génétique médicale |
|---|
| CIM-10 | Q87.3 |
|---|---|
| OMIM | 130650 |
| DiseasesDB | 14141 |
| MedlinePlus | 001186 |
| eMedicine | 919477 |
| MeSH | D001506 |
| GeneReviews | |
| Patient UK | Beckwith-Wiedemann-Syndrome |
![]() Mise en garde médicale
Mise en garde médicale
Causes
Les causes génétiques du syndrome de Beckwith-Wiedemann sont complexes. Dans la majorité des cas on retrouve une expression biallélique du gène IGF-2, localisé sur le Chromosome 11[1], au cours du développement. Cette anomalie est de type épigénétique[2]. D'autres formes ont une méthylation aberrante des gènes H19 ou KCNQ1OT1 situés sur ce même chromosome, les premiers étant plus susceptible de développer des tumeurs[3]. D'autres ont une mutation sur le gène CDKN1C[4], cette dernière étant possiblement responsable des défauts de méthylation décrits[5].
Cette maladie résulte généralement de la régulation anormale des gènes dans une région particulière du chromosome 11. Les gens héritent normalement d'une copie de ce chromosome de chaque parent. Pour la plupart des gènes du chromosome 11, les deux copies du gène sont exprimées ou «activées» dans les cellules. Pour certains gènes, cependant, seule la copie héritée du père d'une personne (la copie héritée paternellement) est exprimée. Pour les autres gènes, seule la copie héritée de la mère d'une personne (la copie héritée de la mère) est exprimée. Ces différences d'expression génique propres aux parents sont causées par un phénomène appelé empreinte génomique. Les anomalies impliquant des gènes sur le chromosome 11 qui subissent une empreinte génomique sont responsables de la plupart des cas de syndrome de Beckwith-Wiedemann.
Au moins la moitié de tous les cas résultent de changements dans un processus appelé méthylation. La méthylation est une réaction chimique qui attache de petites molécules appelées groupes méthyle à certains segments de l'ADN. Dans les gènes qui subissent une empreinte génomique, la méthylation est une façon dont le parent d'origine d'un gène est marqué pendant la formation des ovules et des spermatozoïdes. Le syndrome de Beckwith-Wiedemann est souvent associé à des changements dans les régions d'ADN sur le chromosome 11 appelés centres d'impression (CI). Les CI contrôlent la méthylation de plusieurs gènes impliqués dans la croissance normale, notamment les gènes CDKN1C, H19, IGF2 et KCNQ1OT1. Une méthylation anormale perturbe la régulation de ces gènes, ce qui entraîne une prolifération excessive et les autres caractéristiques du syndrome de Beckwith-Wiedemann.
Environ vingt pour cent des cas de syndrome de Beckwith-Wiedemann sont causés par un changement génétique appelé disomie uniparentale paternelle (UPD). L'UPD paternelle oblige les gens à avoir deux copies actives de gènes hérités paternellement plutôt qu'une copie active du père et une copie inactive de la mère. Les personnes atteintes d'une UPD paternelle sont également dépourvues de gènes qui ne sont actifs que sur la copie héréditaire maternelle du chromosome. Dans le syndrome de Beckwith-Wiedemann, l'UPD paternelle survient généralement au début du développement embryonnaire et n'affecte que certaines cellules du corps. Ce phénomène est appelé mosaïcisme. L'UPD paternelle mosaïque entraîne un déséquilibre des gènes paternels et maternels actifs sur le chromosome 11, qui sous-tend les signes et symptômes de la maladie.
Plus rarement, les mutations du gène CDKN1C provoquent le syndrome de Beckwith-Wiedemann. Ce gène fournit des instructions pour fabriquer une protéine qui aide à contrôler la croissance avant la naissance. Des mutations dans le gène CDKN1C empêchent cette protéine de freiner la croissance, ce qui conduit aux anomalies caractéristiques du syndrome.
Environ 1 % de toutes les personnes atteintes du syndrome de Beckwith-Wiedemann ont une anomalie chromosomique telle qu'un réarrangement (translocation), une copie anormale (duplication) ou une perte (suppression) de matériel génétique du chromosome 11. Comme les autres changements génétiques responsables de Beckwith-Wiedemann, ces anomalies perturbent la régulation normale de certains gènes sur ce chromosome
Assistance médicale à la procréation
L'AMP (Assistance médicale à la procréation) est un terme générique faisant référence aux méthodes utilisées pour obtenir une grossesse par des moyens artificiels ou partiellement artificiels. Les procédures AMP consistent en général à retirer chirurgicalement les ovules des ovaires d'une femme, à les combiner avec du sperme en laboratoire et à les remettre dans le corps de la femme ou à les donner à une autre femme. L'AMP a été associée à des syndromes épigénétiques, en particulier le syndrome de Beckwith-Wiedemann et le syndrome d'Angelman. Trois groupes ont montré un taux accru de conception ART chez les enfants atteints de BWS[6] - [7] - [8] - [9]. Une autre étude a révélé que les enfants conçus par fécondation in vitro (FIV) sont trois à quatre fois plus susceptibles de développer la maladie[10]. Aucun type spécifique d'AMP n'a été associé plus étroitement au syndrome que d'autres, et le mécanisme par lequel l'AMP produirait cet effet n'est pas encore expliqué.
Incidence et prévalence
Le syndrome de Beckwith-Wiedemann affecte 1 nouveau-né sur 10 500 à 13 700 dans le monde[11]. Ce syndrome peut en fait être plus courant que cette estimation car certaines personnes présentant des symptômes bénins ne sont jamais diagnostiquées.
Le SBW a été signalé dans une grande variété de populations avec une incidence égale chez les hommes et les femmes.
Description
Le syndrome de Beckwith-Wiedemann est une affection qui affecte de nombreuses parties du corps. Il est classé comme un syndrome macrosomique , ce qui signifie que les nourrissons touchés sont considérablement plus gros que la normale (macrosomie) et ont tendance à être plus grands que leurs pairs pendant l'enfance. La croissance commence à ralentir vers l'âge de 8 ans, et les adultes atteints de cette maladie ne sont pas inhabituellement grands. Chez certains enfants atteints du syndrome, des parties spécifiques du corps d'un côté ou de l'autre peuvent devenir anormalement grandes, conduisant à une apparence asymétrique ou inégale. Ce schéma de croissance inhabituel, connu sous le nom d'hémi-hyperplasie, devient généralement moins apparent au fil du temps.
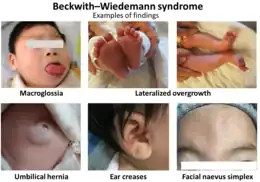
Les signes et symptômes du syndrome varient selon les individus affectés. Certains enfants atteints de cette maladie naissent avec une ouverture dans la paroi de l'abdomen (une omphalocèle) qui permet aux organes abdominaux de dépasser à travers le nombril. D'autres défauts de la paroi abdominale, comme une poche molle autour du nombril (une hernie ombilicale), sont également fréquents. Certains nourrissons atteints du syndrome de Beckwith-Wiedemann ont une langue anormalement grande (macroglossie), ce qui peut gêner la respiration, la déglutition et la parole. D'autres caractéristiques majeures de cette affection comprennent des organes abdominaux anormalement grands (viscéromégalie), des plis ou des piqûres dans la peau près des oreilles, une glycémie basse (hypoglycémie) pendant la petite enfance et des anomalies rénales.
Les enfants atteints par le syndrome ont un risque accru de développer plusieurs types de tumeurs cancéreuses et non cancéreuses, en particulier une forme de cancer du rein appelée tumeur de Wilms et une forme de cancer du foie appelée hépatoblastome. Les tumeurs se développent chez environ 10% des personnes atteintes de cette maladie et apparaissent presque toujours dans l'enfance.
La plupart des enfants et des adultes atteints du syndrome n'ont cependant pas de problèmes médicaux graves associés à la maladie. Leur espérance de vie est généralement normale.
Évolution
Le risque de survenue d'un cancer est important dans les premières années de la vie, surtout à type de neuroblastome, néphroblastome ou d'hépatoblastome[12] - [13].
Prise en charge et traitement
Prise en charge
La prise en charge implique des stratégies médicales palliatives, chirurgicales. La surveillance tumorale doit être instaurée (chez le patient suspect ou diagnostiqué, ainsi que chez le jumeau asymptomatique). celle-ci ne doit pas être guidée par des corrélations génotype/phénotype. Le dépistage néonatal d'une hypoglycémie doit être entrepris s'il existe des signes prénataux diagnostiques ou évocateurs ou même chez le nouveau-né asymptomatique mais à haut risque du fait d'antécédents familiaux[14].
Diagnostic prénatal
Le diagnostic génétique peut être fait par prélèvement invasif (biopsie de trophoblaste ou amniocentèse): réalisation d’un caryotype et d’une puce à ADN (ACPA). Le plus souvent. Le caryotype est normal. L’étude en génétique moléculaire peut permettre de mettre en évidence une anomalie de la région 11p15.5 (sur le bras court du chromosome 11). A noter qu’un tiers dès Wiedemann- Beckwith n’ont pas d’anomalie génétique identifiée.
Notes et références
- Murrell A, et al. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: interaction between genotype and epigenotype, Hum Mol Genet, 2004;13:247–255
- Choufani S, Shuman C, Weksberg R, Molecular findings in Beckwith-Wiedemann syndrome, Am J Med Genet C Semin Med Genet, 2013;163C:131–140
- Bliek J, Maas SM, Ruijter JM et al. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS, Hum Mol Genet, 2001;10:467–476
- Lam WW, Hatada I, Ohishi S et al. Analysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype-phenotype correlation, J Med Genet, 1999;36:518–523
- Li M, Squire J, Shuman C et al. Imprinting status of 11p15 genes in Beckwith-Wiedemann syndrome patients with CDKN1C mutations, Genomics, 2001;74:370–376
- DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP, « Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith–Wiedemann syndrome with cancer and birth defects », American Journal of Human Genetics, vol. 70, no 3, , p. 604–11 (PMID 11813134, PMCID 384940, DOI 10.1086/338934)
- Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y, « In vitro fertilization may increase the risk of Beckwith–Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene », American Journal of Human Genetics, vol. 72, no 5, , p. 1338–41 (PMID 12772698, PMCID 1180288, DOI 10.1086/374824)
- Maher ER, Brueton LA, Bowdin SC et al., « Beckwith–Wiedemann syndrome and assisted reproduction technology (ART) », Journal of Medical Genetics, vol. 40, no 1, , p. 62–4 (PMID 12525545, PMCID 1735252, DOI 10.1136/jmg.40.1.62)
- Chang AS, Moley KH, Wangler M, Feinberg AP, Debaun MR, « Association between Beckwith–Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients », Fertility and Sterility, vol. 83, no 2, , p. 349–54 (PMID 15705373, PMCID 4872595, DOI 10.1016/j.fertnstert.2004.07.964)
- Gosden R, Trasler J, Lucifero D, Faddy M, « Rare congenital disorders, imprinted genes, and assisted reproductive technology », Lancet, vol. 361, no 9373, , p. 1975–7 (PMID 12801753, DOI 10.1016/S0140-6736(03)13592-1, S2CID 9442967)
- Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, Richiardi L, Larizza L, Silengo MC, Riccio A, Ferrero GB. Prevalence of Beckwith-Wiedemann syndrome in North West of Italy. Am J Med Genet A. 2013;161A:2481–6.
- DeBaun MR, Tucker MA, Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry, J Pediatr, 1998;132:398–400
- Logan G. Spector et Jill Birch, « The epidemiology of hepatoblastoma », Pediatric Blood & Cancer, vol. 59, no 5, , p. 776–779 (ISSN 1545-5017, PMID 22692949, DOI 10.1002/pbc.24215, lire en ligne, consulté le )
- Orphanet,
Liens externes
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number:130650
- (en) GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1993-2005
- Site orphanet