Spectroscopie d'absorption atomique par four graphite
La spectrométrie d’absorption atomique par four graphite (GFAAS), aussi nommée spectrométrie d’absorption atomique électrothermique (ETAAS)[1] est une technique analytique utilisée pour l'analyse d'éléments chimiques[2]. Elle a été développée dans les années 1950 par Boris V. L’Vov[3]. Cette méthode est utilisée pour l’étude qualitative et quantitative de presque tous les métaux, les métalloïdes et certains non métaux[2]. Comme dans toutes les techniques de spectrométrie d'absorption atomique, la GFAAS contient les parties suivantes : une source de raies (lampe) qui émet un rayonnement linéaire de résonance, un atomiseur (ou chambre d’atomisation), soit un tube graphite où il se produit la vaporisation de l’analyte, un monochromateur qui sélectionne la bonne longueur d’onde selon l’analyte observé (rayons UV ou visibles), un détecteur qui mesure la quantité d’absorption et un système informatique pour traiter les informations provenant de l’analyse[4].
Principes théoriques
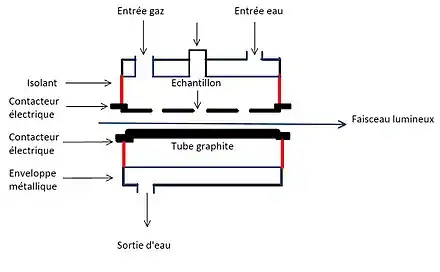
Généralement, les atomiseurs ii présents en spectroscopie d’absorption atomique utilisent comme source d’énergie une flamme. La GFAAS est une méthode dite « sans flamme » mettant en jeu des tubes ou baguettes graphites chauffées électriquement. Le four est composé d’un cylindre creux en graphite avec des dimensions pouvant varier de 20 à 50 mm pour la longueur et de 3 à 9 mm pour le diamètre intérieur[2] - [6]. Ce cylindre est placé de sorte à avoir le faisceau incident dans l’axe du tube[6]. Celui-ci est entouré d’une enveloppe métallique où l’eau circule[6]. Un espace sépare le tube et son enveloppe dans lequel est envoyé un flux de gaz inerte[6]. L’argon est généralement utilisé afin d’exclure l’oxygène et d’empêcher la combustion du graphite[2]. Le gaz passe aussi au travers du tube par le biais d’orifices conçus à cet effet[6]. Lorsque débute l’analyse, le flux d’argon est désactivé afin de maximiser le temps pendant lequel l'analyte se trouve dans le tube et donc la sensibilité[3]. En plus de l’argon, des fenêtres en quartz isolantes sont placées de chaque côté du four afin d'empêcher l'entrée d'oxygène par le biais du chemin optique[2]. Les contacteurs électriques correspondent à des électrodes. Celles-ci sont connectées à une source d'alimentation en courant alternatif basse tension à courant élevé qui sert à chauffer le tube de graphite[2]. Ce système permet le passage de la température ambiante à 2 700 °C[2]. L’introduction de l’échantillon est effectuée au niveau du centre du tube de graphite au niveau des orifices de l’enveloppe et du tube[6]. Cette région peut atteindre la température nominale d'atomisation prédéfinie[7]. Cette méthode analytique est habituellement utilisée pour des échantillons liquides, mais elle peut aussi analyser des échantillons solides[7]. Pour les liquides, l'introduction de l'échantillon s'effectue à l’aide d’une micropipette. Typiquement, le volume est d’environ 20 µL[2], mais celui-ci peut varier de 1 à 100 µL[6]. Ce volume est ajouté précisément à l’aide d’un échantillonneur automatique[2]. Concernant les solides, la masse d'échantillon analysée doit être comprise entre 0,1 et 1 mg[7]. En ce qui concerne l'introduction d'échantillons solides dans le four graphite, différents systèmes sont maintenant disponibles dans le commerce. La société allemande Jena a lancé un appareil qui introduit automatiquement les plates-formes contenant les échantillons solides dans le tube de graphite[8].
Chaque analyse d’absorption électrothermique (ETAAS) utilise un programme d’essai configuré après l’introduction de l’échantillon. L'objectif principal de toutes les méthodes qui utilisent un four graphite est d'obtenir une atomisation / vaporisation sélective de l'analyte. Pour ce faire, il est essentiel d'optimiser un programme de température[8].
Les différentes étapes du programme du four sont les suivantes[2] :
- le séchage pour éliminer l'eau de l'échantillon (100 à 150 °C) ;
- la pyrolyse pour éliminer les matières volatiles de la matrice d'échantillon (300 à 1 500 °C) ;
- le refroidissement pour permettre au four d’atteindre une température relativement basse (200 °C) ;
- l’atomisation dans laquelle l'analyte est convertie en atomes permettant la mesure analytique (1 600 à 2 700 °C) ;
- la dernière étape est appelée l’étape « propre » pour éliminer les matières résiduelles du four (2 500 à 2 700 °C).
L’atomisation est l’étape primordiale dans l’analyse de l’échantillon. Elle est définie comme la conversion de l’échantillon en atomes gazeux qui absorbent le rayonnement[1]. Elle se produit grâce au réchauffement rapide du four, car cela permet de minimiser les interférences[2]. Afin de conserver la chaleur dans le tube, une petite étagère (plate-forme L'vov) est insérée à l'intérieur de celui-ci[2], assurant que l'atomisation se produit dans un environnement chaud[2].
Après l’étape d’atomisation, le faisceau lumineux traverse le tube et le système de détection pour l’AAS[2]. Les raies d’absorption obtenues par cette méthode peuvent durer plusieurs secondes avant d'être enregistrées[9]. L'absorption est mesurée au-dessus de la surface chauffée où l'échantillon a été atomisé[1], grâce à un monochromateur. Il existe différents monochromateurs. Celui étant le plus couramment utilisé en GFAAS est le monochromateur à réseau[1]. Il possède cinq composants : une fente d'entrée, une lentille de collimation ou un miroir, un réseau de diffraction, un élément de focalisation et une fente de sortie[1]. Le réseau reflète et disperse les longueurs d'onde des composants dans un miroir qui focalise une bande étroite de longueurs d'onde sur une fente de sortie[1]. Les largeurs des fentes sont importantes quant à la sélection de la longueur d’onde étudiée[1]. Elles doivent être aussi étroites que possible tout en laissant suffisamment de puissance rayonnante pour atteindre le détecteur[1]. Si les fentes sont trop larges, plusieurs longueurs d'onde traverseront et causeront une mauvaise résolution[1]. Si les fentes sont trop étroites, la puissance radiante qui peut atteindre le détecteur sera réduite et difficile à détecter[1]. Par conséquent, la largeur de la fente est un compromis entre la résolution et la détectabilité. Les réseaux peuvent réfléchir et disperser les rayons ultraviolets collimatés, visibles et infrarouges[1].
Une fois passé dans le monochromateur, le faisceau lumineux atteint le détecteur. Dans la spectrométrie d'absorption atomique par four graphite, la quantité de rayonnement qui traverse un échantillon est mesurée et quantifiée par transmittance[1]. Lorsque la lumière traverse un échantillon, la puissance est atténuée, car elle est absorbée par l'analyte dans l'échantillon. Le détecteur le plus utilisé en GFAAS est le tube multiplicateur[1].
Un tube photomultiplicateur (PMT) est utilisé pour la mesure de la puissance radiante faible[1]. Contrairement à un phototube traditionnel, le PMT contient des électrodes supplémentaires appelées dynodes[1]. En raison de la charge positive croissante, les électrons sont accélérés vers les dynodes. Après avoir traversé les dynodes, les électrons se rassemblent à l'anode où ils sont recueillis sous la forme d'un courant[1]. Ce courant est ensuite converti en une tension et mesuré. Les PMT se limitent à des mesures de sources de rayonnement de faible puissance[1].
Voie d'amélioration de la technique pour un échantillon solide
Voir[8].
Depuis les années 1980, la détermination directe d'éléments à partir d'échantillons solides par AAS a été le plus souvent réalisée à l'aide d'un four graphite (graphite furnace, GF)[8].
De nos jours, il est bien évident que la production directe d'un signal analytique à partir d'un échantillon solide offre un certain nombre d'avantages importants résultant de l'élimination de l'étape de dissolution[8] :
- le risque de contamination est considérablement réduit, ainsi que le risque de pertes d'analyte ;
- la sensibilité augmente, car les échantillons ne sont pas dilués ;
- les résultats sont obtenus rapidement ;
- une petite quantité d’échantillon suffit, dans la plupart des cas, à faire l’analyse ;
- l'utilisation de réactifs corrosifs ou dangereux n'est pas nécessaire, ce qui entraîne des avantages économiques et environnementaux.
Afin d'optimiser l'atomisation de l'analyte, il faut avant tout s'intéresser à la programmation de la température. Cette optimisation peut cependant être plus difficile à atteindre avec des échantillons solides pour deux raisons principales. La première est que, très souvent, lors du processus de dissolution de l'échantillon ou dans un traitement ultérieur, une partie significative de la matrice est éliminée (par exemple, la matière organique peut être détruite, les silicates peuvent être éliminés, etc.)[8]. De cette façon, l'aliquote de l'échantillon dissout introduit dans le GF contient moins de composés que l'échantillon original et ceux-ci sont moins concentrés que dans l'échantillon d'origine[8]. La seconde raison est que le processus de dissolution de l'échantillon implique également la séparation de l'analyte de la matrice, de sorte qu'il est normalement introduit dans le GF comme une espèce chimique simple (habituellement un cation ou un complexe soluble)[8].
Comme il l'a été annoncé précédemment, travailler directement avec des échantillons solides implique la présence de la matrice entière dans l'atomiseur et ceci pose donc des problèmes qui sont particuliers à cette procédure[8]. La vaporisation simultanée de certains composés de la matrice avec l'analyte peut entraîner, pour le GFAAS, une augmentation du signal de fond (absorption moléculaire, diffusion de la lumière par des particules solides condensées, etc.) ainsi qu'un risque accru d'interférences dans la phase gazeuse[8]. De plus, il est possible que le processus d'atomisation d'un échantillon solide diffère de celui qui se produit lorsque l'analyte est introduit en solution[8].
Les méthodes possibles pour obtenir une atomisation sélective de l'analyte en fonction de la relation entre les volatilités relatives de l'analyte et de la matrice sont décrites ci-dessous[8].
Si la matrice est plus volatile que l'analyte, l'étape de pyrolyse devient la plus importante, car il faut éliminer autant de matrice que possible pendant cette étape. Elle peut être ainsi considérée comme une microdigestion / incinération, mais généralement, la température d'atomisation n'est pas très cruciale[8]. Ce cas-ci est celui présentant le moins de difficultés lors d'analyse. Il correspond, par exemple, à la détermination de la plupart des métaux dans les matrices organiques[8].
Si l'analyte est plus volatil que la matrice, l'étape de pyrolyse est moins importante et peut même être omise[8]. Cependant, l'étape d'atomisation devient essentielle, car la température doit être la plus basse possible pour permettre une atomisation maximale de l'analyte et une vaporisation minimale de la matrice[8]. Ce cas est rencontré lors de l'analyse de certains échantillons inorganiques, lorsque l'analyte est volatil (par exemple le cadmium, le mercure) ou lorsque la matrice est réfractaire[8].
La situation la plus compliquée est rencontrée lorsque la température d'atomisation de l'analyte et la température de vaporisation de la matrice sont similaires, comme cela se produit dans la détermination des analytes volatils dans les matrices organiques et dans la majorité des déterminations avec des matrices inorganiques[8]. En conséquence, la solution la plus générale semble être l'utilisation de modificateurs chimiques. Le rôle des modificateurs chimiques est d'interagir avec l'analyte ou avec la matrice en modifiant sélectivement sa volatilité[8]. Le modificateur chimique peut être ajouté sous forme de solution, de gaz ou même de solide. L'un des défis consiste toujours à développer un programme de température approprié pour assurer une interaction efficace entre les modificateurs et l'élément cible, à l'origine intégré dans le solide[8]. Pour les matrices organiques, l'approche la plus largement utilisée implique un modificateur, tel que le palladium, qui interagit avec l'analyte pour permettre des températures de pyrolyse plus élevées sans perte d'analyte. Les agents de fluoration peuvent être utilisés pour améliorer la volatilité de la matrice par rapport à l'analyte ou vice versa. Dans le cas des matrices réfractaires, l'utilisation de l'hydrogène a été proposée pour éviter la formation d'oxydes volatils en veillant à ce qu'une quantité significative de la matrice soit vaporisée avec les analytes[8]. L'ajout de poudre de graphite s'est avéré bénéfique, en particulier pour les silicates, en obtenant pics unimodaux[8].
Applications
La GFAAS est utilisée dans le domaine du pétrole. Les métaux comme le V et Ni présents dans les pétroles bruts peuvent être détectés, et le Pb, Sb, As, Cd, Cr, Co, Mn et Mo dans les produits pétroliers[10].
Elle est aussi utilisée dans le domaine environnemental[11] :
- elle peut être configurée avec un système de ventilation afin de capturer les gaz toxiques et corrosifs émis par la vaporisation des échantillons aliquotes ;
- elle permet de déterminer et de séparer les quantités de Ni et Co présentes à la surface des eaux de mer ;
- la GFAAS est utilisée à la préparation et caractérisations de nanoparticules magnétiques pour la détermination de l'Au, Pd et Pt dans les échantillons miniers ;
- elle peut être également utilisée dans l'analyse du Pt provenant des gaz d'échappement des voitures[7].
Elle est suffisamment sensible pour la détermination des métaux nobles dans les échantillons géologiques et pour la surveillance directe des médicaments anticancéreux à base de Pt dans les fluides biologiques et les tissus[12].
La détermination des contaminants inorganiques dans les cosmétiques faciaux peut être analysée par cette technique analytique[13].
La GFAAS peut aussi permettre l’analyse de matériaux afin d’y identifier les impuretés (ex. : présence de Co, Fe Ni et Pb dans les nanotubes de carbone)[14].
Avantages et inconvénients
Avantages
- La GFAAS peut analyser de très faibles quantités d'échantillon (jusqu'à 0,5 µL)[9]. Ainsi, l’exposition aux produits chimiques toxiques est minimisée[6].
- Il n'y a peu (ou pas) d'erreurs sur la préparation de l'échantillon, car certains échantillons solides ne nécessitent pas de dissolution préalable[9].
- Cette technique est très sensible avec une limite de détection de l’ordre de 0,1 µg/L pour la plupart des éléments[6]. Cela correspond à une limite de détection cent à mille fois plus faible par rapport à la spectrométrie d'absorption de flamme[9]. Cela est dû à une augmentation du temps de séjour des vapeurs d'échantillon dans le trajet optique[7]. Cette sensibilité permet l’analyse d’ultra-traces dans les échantillons[8].
- La GFAAS est plus précise que les méthodes utilisant une flamme[9].
- Bien que la GFAAS soit généralement appelée une technique à élément unique, les instruments les plus récents possèdent une capacité multi-éléments simultanée pour jusqu'à six analytes[7].
- Le coût de l'instrument est raisonnable[8].
Inconvénients
- Cette technique a généralement plus de bruits de fond[9].
- La minéralisation peut induire une perte d'analyte de certains composés comme l'arsenic, le mercure et sélénium qui sont des éléments volatils[9].
- Le four graphite ne peut parfois pas complètement atomiser l'échantillon, ce qui provoque des effets « mémoire » dans celui-ci[9].
- Sa sensibilité est le facteur affectant les limites de détection dans le cas d'un échantillon contaminé[2].
- Un seul élément à la fois est analysé, ce qui engendre un faible débit d'échantillons[2].
- Le temps d'atomisation est plus important qu'un atomiseur à flamme, car le temps de chauffe est long[10].
- Chaque tube peut être utilisé pour 100 à 200 analyses selon la nature des éléments à doser[9].
- L'étalonnage d'un échantillon solide est difficile à obtenir avec l'appareil, car l'atomisation de l'analyte peut être influencée par la matrice utilisée[8].
Notes et références
- (en) « Atomic Absorption Spectroscopy Learning Module », sur maryville.edu, (consulté le )
- (en) David J. Butcher, « Advances in electrothermal atomization for atomic absorption and atomic fluorescence », Applied Spectroscopy Reviews, , p. 305-319 (ISSN 0570-4928, lire en ligne)
- (en) Maurice Pinta, Atomic Absorption Spectrometry, Londres, 418 p., p. 69-70
- Skoog D. A., West D. M., Holler F. J. et Crouch S. R. (trad. de l'anglais), Chimie analytique, Bruxelles, De Boeck, , 1116 p. (ISBN 978-2-8041-9071-2, lire en ligne)
- J. Mendham, R. C. Denney, J.D. Barnes, M. Tomas (trad. de l'anglais), Analyse Chimique Quantitative de Vogel, Bruxelles, de broeck, , 889 p. (ISBN 2-8041-4799-1), p. 645-646
- (en) G. Schlemmer, Analytical Graphite Furnace Atomic Absorption Spectrometry : A Laboratory Guide, Suisse, Birkhaüser, , 285 p. (ISBN 3-7643-5770-3), p. 35-36
- (en) L. Bencs et al., « Methods for the determination of platinum group elements originating from the abrasion of automotive catalytic converters », Spectrochimica Acta Part B, , p. 1723–1755
- (en) M.A. Belarra, M. Resano, F. Vanhaecke et L. Moens, « Direct solid sampling with electrothermal vaporization/atomization: what for and how? », Trends in Analytical Chemistry, , p. 828-839
- J. Mendham, R. C. Denney, J. D. Barnes et M. Thomas (trad. de l'anglais), Analyse chimique quantitative de Vogel, Bruxelles, de broeck, , 889 p. (ISBN 2-8041-4799-1), p. 645-646
- (en) R.A. Nadkarni, Modern instrumental methods of elemental analysis of petroleum products and lubricants, Philadelphie, ASTM, , 154 p. (ISBN 0-8031-1416-8, lire en ligne), p. 23
- (en) « Sample records for absorption spectrophotometry gfaas », sur www.science.gov (consulté le )
- (en) Beata Godlewska-żyłkiewicz, « Preconcentration and Separation Procedures for the Spectrochemical Determination of Platinum and Palladium », Microchim. Acta, , p. 189–210
- (en) Ariane I. Barros, Tiago V. Silva, Edilene C. Ferreira et José A. Gomes Neto, « Determination of Lead in Eye Shadow and Blush by High-Resolution Continuum Source Graphite Furnace Atomic Absorption Spectrometry Employing Direct Solid Sampling », Journal of the Brazilian Chemical Society, , p. 140-146 (lire en ligne)
- (en) Martin Resano, « Simultaneous determination of Co, Fe, Ni and Pb in carbon nanotubes by means of solid sampling highresolution continuum source graphite furnace atomic absorption spectrometry », Journal of Analytical Atomic Spectrometry, , p. 657 (ISSN 0267-9477)
Voir aussi
Bibliographie
- (en) Michelle Gende et Martina Schmeling, « Development of an analytical method for determination of lead and cadmium in biological materials by GFAAS using Escherichia coli as model substance », PLOS One, vol. 17, no 5, , e0267775 (ISSN 1932-6203, DOI 10.1371/journal.pone.0267775, lire en ligne, consulté le ).